
Describing and classifying variants

Before we look at the processes bioinformaticians go through to filter and analyse variants, let’s stop and look at variants and how we name and classify them.
The importance of classifiying variants
The consistent naming and use of the correct nomenclature when referring to variants is extremely important in Mendelian inherited diseases and even more so for particularly rare diseases. As we will see establishment of pathogenicity does rely on searching the scientific literature for evidence of pathogenicity to associate genotype with experimentally verified disease association and also databases of clinically significant variants, thus this process is aided significantly if variants are referred to in a consistent manner.
Naming sequence variants
The Human Genome Variation Society provides guidance on how to name sequence variants 1, all variants should be reported with reference to the gene or reference sequence that it has been found in, for example, for the gene responsible for cystic fibrosis : (CFTR; NM_000492.3),
-
CFTR is the gene name which is described in italics
-
NM_000492.3 is the accession number reference for the gene in the NCBI database, the most common variant causing cystic fibrosis is p.Phe508del
-
p – defining the protein sequence, and,
-
Phe508del – denotating a deletion of a phenylalanine at position 508 in the protein sequence.
The HGVS recommends naming variants according to the genomic sequence therefore there is a handy tool called mutalyzer 2 that will convert between different descriptors of variants. The full variant will therefore be referred to as NM_000492.3(CFTR):c.1521_1523delCTT.
Increasing numbers of variants
With the clinical application of gene panel (whole exome and now whole genome testing) increasing numbers of variants are being returned, and importantly growing numbers of variants of uncertain or unknown significance VOUS are being returned. This is a variant for which there is no substantive published evidence of functional effect or link with pathogenicity even though it may be in a gene where other variants that have been linked to pathogenicity have been identified.
Clinical reporting of the likely pathogenicity of a variant in Mendelian inherited diseases helps the clinician in clinical decision making, e.g. in some cases the disease may be treatable so this will inform treatment options, in other cases it may help to inform the family with regards to pre-natal testing or testing of other family members. Therefore it is important that interpretation of variants is done within the context of the presenting phenotype and other information that has already been established regarding that patient. There could be many rare or de novo variants found when sequencing an individual, particularly when using gene panels, therefore the important step in the interpretation is establishing which variant is most likely to be responsible for the condition, looking at all lines of evidence. The American College of Medical Genetics and Genomics has developed guidelines and a standard terminology to describe variants identified in genes that cause Mendelian disorders. 3 This system defines 5 classes 1. Benign, 2. Likely benign, 3. Uncertain significance , 4. Likely pathogenic and 5. Pathogenic.
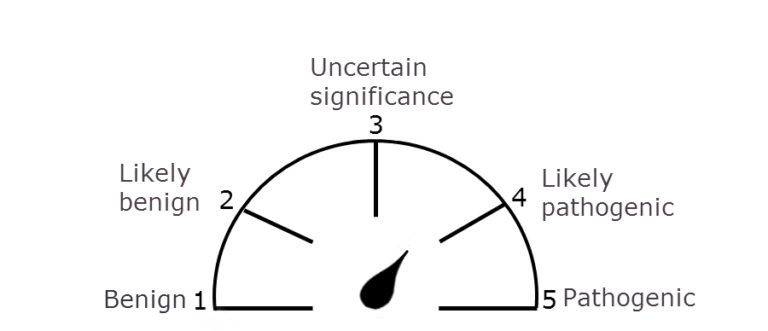
Very strong evidence of pathogenicity would be supported by a variant that causes the production of a non-sense protein or a truncated protein where loss of function is associated with pathogenicity. Strong evidence would be provided by either in-vitro or in-vivo functional studies supportive of a damaging effect on the gene or gene product. A variant that is likely pathogenic combined with other clinical evidence would be sufficient to inform clinical decision making and management of that patient, however a variant of uncertain significance would not be.
Re-analysis of variants
The availability of new population frequency data such as from the ExAC project has allowed some variants that were of uncertain significance to be re-classified as benign, or for example new family data may also bring new evidence to light.
The important point here is that with the increasing usage of whole exome sequencing and gene panels there are growing numbers of variants of uncertain significance are being found. Therefore, these variants need to be recorded and documented in a way that allows them to be searchable and readily available for reanalysis.
Over to you!
- How should we report variants of unknown significance to the clinician?
- Should there be a process for the re-analysis of variants in light of new information?
- Why and how should we store or share variant data?
References
-
Mutation nomenclature extensions and suggestions to describe comples mutations: a discussion by den Dunnen JT and Antonarakis SE (2000) in Hum.Mutat. 15:7-12
-
Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. by Wildeman M et al. (2008) in Hum Mutat 29, 6-13
-
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Genetics in Medicine 17;5 405-424 doi:10.1038/gim.2015.30
Clinical Bioinformatics: Unlocking Genomics in Healthcare


Clinical Bioinformatics: Unlocking Genomics in Healthcare
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




