
Molecular typing methods for outbreak investigation

To understand outbreaks of disease, it is essential to know whether or not different cases are caused by the same infectious agent.
Many different typing methods are available to do this – whole-genome sequencing can be thought of as an extremely detailed kind of typing. At the simpler end are methods that identify some features of the bacterium without using any DNA sequence information. For example, Gram staining (discussed in week 1) is based on whether a specific dye, crystal violet, sticks to a bacterium, and can be used to classify bacteria into Gram-positive (e.g. S. aureus) and Gram-negative (e.g. E. coli). Serotyping classifies bacteria of the same species based on molecules on the cell surface; the E. coli strains in the outbreak we have just discussed were identified by serotyping. Antibiotic resistance profiling classifies bacteria based on the antibiotics to which they are resistant.
There are a variety of DNA-based typing techniques that do not involve whole genome sequencing. These have been used because they have been cheaper and easier than whole genome sequencing.
Variable-number tandem repeat (VNTR) typing
Bacterial genomes contain many repetitive elements. These often consist of fairly short nucleotide repeats (the example below is 53bp long) that can change in number because of errors in DNA replication. VNTR typing takes advantage of this variability. The size of these elements, rather than their sequence, is determined in each strain of the bacterium under investigation. More similar strains will have repeats with more similar lengths.
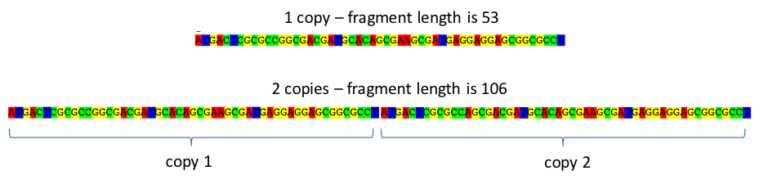
Example of a VNTR locus in M. tuberculosis (Click image to expand) The number of copies determines the fragment length, which can be distinguished by observing the banding pattern using gel electrophoresis.
Pulsed field gel electrophoresis (PFGE) typing
PFGE typing involves cutting bacterial genomic DNA into large fragments using a restriction enzyme. This is a molecular tool that cuts DNA at a specific sequence, known as a restriction site. The size of the fragments can differ between bacterial strains because changes in the genome will affect the number of restriction sites and the spaces between them. The lengths of the DNA fragments are measured by ‘pulsed-field’ gel electrophoresis. This provides a distinctive pattern of fragment sizes for each bacterial strain.
Multi-locus sequence typing (MLST)
MLST involves sequencing regions of 400-500bp from multiple (usually seven) genes. These are usually essential genes, present in all strains. Each unique sequence for each gene is assigned an arbitrary number, and is combined to generate a code called a sequence type (ST). One of the main advantages of MLST is that it is highly reproducible and nucleotide sequences are easily comparable between laboratories. Large databases of ST profiles for different species are available (https://pubmlst.org/).
Whole genome sequencing
Whole genome sequencing is rapidly becoming cheap enough to be used as a routine typing tool. Instead of using specific parts of the genome, the entire genome can be used to distinguish strains. This is primarily done by identifying single nucleotide differences between each strain and the assembled genome of a reference strain (resequencing). This provides much greater resolution to distinguish closely related strains and picks up differences that would be missed by VNTR, PFGE or MLST typing. In the next video we will be talking to Julian Parkhill who will tell us more about the advantages of whole genome sequencing for transmission tracking.
Bacterial Genomes: Disease Outbreaks and Antimicrobial Resistance


Bacterial Genomes: Disease Outbreaks and Antimicrobial Resistance
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




