
How do we find polygenic genes?
You’ve learnt about the genetic architecture of type 1 and type 2 diabetes, but how have we made these discoveries and how do we go about finding new genes involved? Before 2007 the process was very much a ‘fishing expedition’, scientists had to predict which genes might be involved based on their knowledge of the causes of diabetes and then test those genes in a series of patients. Around 2007 there was a substantial improvement in genomic technology that allowed scientists to test genetic variants across the whole genome in a single experiment at a reasonable cost. Such tests now cost approximately $50 per patient.
The method makes use of the phenomenon of linkage disequilibrium, which means that genetic variants that are physically close to each other and not separated by recombination during meiosis will be inherited together. So for example, if variants are on different chromosomes they can only be passed on together by chance, there is a 50% chance that those two variants will appear together in the same sperm or egg cell (gamete). If variants are within a few bases of each other they will almost always be inherited together, so a 100% chance of them being in a gamete together. The chance that they will be together diminishes as the distance between them increases. This means that by testing one genetic variant it gives you information about the genetic variation nearby. Thus by testing variants spread across the genome, you obtain information about variation over a large proportion of the genome.
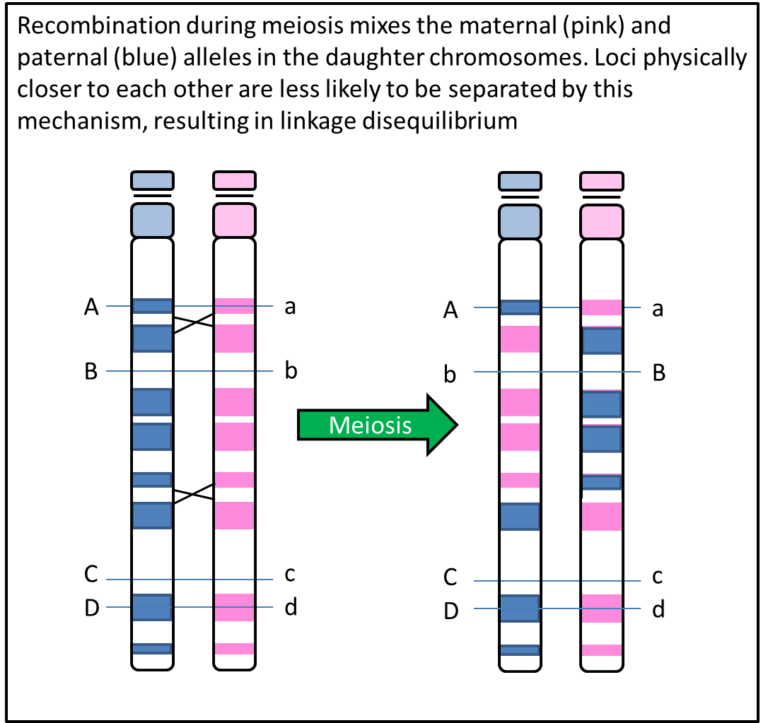
This principle is exploited in Genome-Wide Association Studies (GWAS). Typically around 500,000 variants are directly tested which capture information about several million untested variants, spanning the 20,000 genes in the genome. The variants tested are Single Nucleotide Polymorphisms (SNPs) which are simple variants that differ at a single DNA nucleotide position with generally two alternative bases or alleles and are the most abundant variants in the genome. In GWAS for Type 1 diabetes and Type 2 diabetes, case control (discontinuous phenotype) studies are used and for each SNP the frequency of each allele of a SNP is compared between cases and controls. For continuous phenotypes, like fasting glucose measures, the mean level for each genotype category is compared.
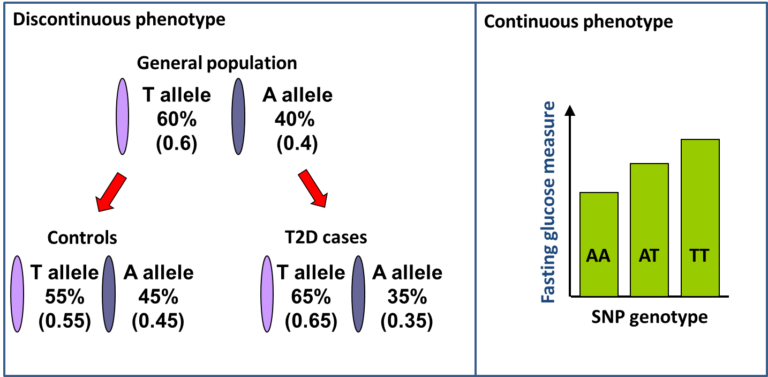
Statistical tests are used to determine whether the difference between cases and controls, or the difference in mean for each genotype, is greater than would be expected by chance. A similar test is applied to all the SNPs across the genome. If the strength of the statistical association with having diabetes passes a threshold, we can be confident that a new genetic region for the disease has been found. GWAS have found around 90 genetic regions that are associated with Type 2 diabetes.
By identifying genetic regions associated with traits and diseases we can understand more about the biology of those traits, perhaps develop novel therapeutics and start to be able predict individuals at increased risk of developing a disease.
Genomic Medicine: Transforming Patient Care in Diabetes


Genomic Medicine: Transforming Patient Care in Diabetes
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




