
What is a sequencing library and how is it prepared?
Next-generation sequencing has revolutionised genomic research, allowing high-throughput profiling of genomes, transcriptomes, DNA-protein interactions and more. This is achieved by performing millions to billions of massively parallel short read (50-500bp) reactions in each run. In order for this to happen, each reaction must first be localised to a solid substrate, such as a bead or a glass slide, before being clonally amplified and sequenced in-situ. On Illumina systems, this process is referred to as clustering.
Before sequencing can begin, DNA or RNA samples must first be converted into molecules compatible with the sequencing platform of choice. We refer to this converted sample as a library, and the processes used to generate it are referred to as library preparation. Optional steps can be added before, during and after library preparation in order to enrich, select, deplete or convert the sample for specific content prior to sequencing (for example to enrich for pathogen DNA over host DNA).
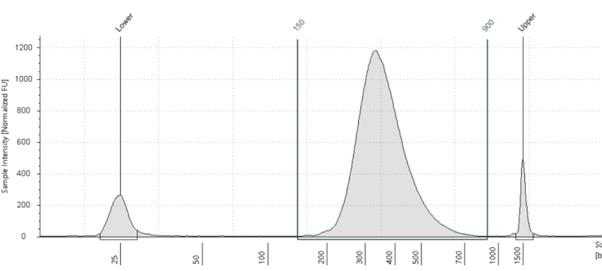
Figure 1 – Electropherogram of a typical NGS library, showing products between 200 and 500bp. Peaks at 25bp and 1500bp are the lower and upper calibration markers.
If the input is RNA, such as for the SARS-CoV-2 virus genome, it must first be converted to DNA – a process known as reverse transcription. From here, there are three common steps in library preparation, summarised as follows:
1. Fragmentation and end-repair
Short read sequences, such as Illumina, do not read long DNA fragments, so molecules must be fragmented to typical lengths of 100-300bp. Fragmentation of DNA can be performed physically, chemically or enzymatically. After fragmentation, these molecules require blunting, (so that each strand is of equal length with no unpaired bases), phosphorylation and A-tailing. Phosphorylation and A-tailing allow for T-tailed adapters to be ligated at the next step.
2. Adapter ligation
Synthetic DNA molecules, known as adapters, are attached to the ends of each library molecule. These adapters have multiple functions, containing domains to allow each molecule to be immobilised and amplified, along with domains for sequencing primer binding and domains containing sample-specific indexes. The use of sample indexes allows for many samples to be pooled prior to sequencing, thus maximising the output of each sequencing run. This is known as multiplex sequencing.
3. PCR amplification (optional)
PCR can be used to select library molecules that have successfully ligated adapters at each end, whilst boosting library yield for sequencing. P5/P7 and index sequences can also be added at this step, if not added at the adapter ligation step. PCR can cause bias in sequencing libraries as GC-rich sequences do not amplify well. If DNA input is sufficiently high, this step can be skipped, reducing PCR bias.

Figure 2 – Schematic of a typical library molecule (Illumina). P5 and P7 are complementary to DNA sequences found attached to the surface of the flow cell, allowing for each molecule to be captured and amplified. Index 1 and Index 2 are sample-specific indexes that allow for multiple samples to be pooled prior to sequencing. By reading each index, each DNA insert can be assigned to its sample of origin at the end of the sequencing run.
4. Tagmentation
An alternative method to the more traditional fragmentation and adapter ligation is a process known as tagmentation whereby a transposome (transposase enzyme plus DNA adapter sequence) simultaneously fragments and adapter tails DNA. Transposases are enzymes that mediate the movement of DNA within a genome (sometimes called a “jumping gene”). Tagmentation is much more efficient than standard fragmentation and ligation, allowing for more sensitive applications that typically have less DNA input. The tagmentation process has recently been simplified by attaching these transposomes to magnetic beads, thus allowing for fixed amounts of DNA to be captured, whilst undergoing uniform fragmentation and adapter addition. This removes the requirement for each library to be quantified before sequencing, reducing turnaround times and hands-on steps whilst facilitating automation.

Figure 3 – Bead linked transposomes simultaneously capture a fixed amount of DNA and fragment to a uniform length, whilst adding Illumina adapter sequences. Source: Illumina
A Practical Guide for SARS-CoV-2 Whole Genome Sequencing


A Practical Guide for SARS-CoV-2 Whole Genome Sequencing
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




