
Assembly of SARS-CoV-2 genome and sequence alignment
Did you ever ask someone what a new delicacy tastes like, and they respond with: “It tastes like chicken.”
The truth is, that we use references all the time. It gives us a place to start. A “reference” point to narrow things down to. When you ask for directions, the person giving you directions will first check whether you are familiar with certain landmarks. We do the same when it comes to sequencing reads.
De novo versus reference genome assembly
When your reads are obtained from the sequencing machine, here are two methods of assembling your reads. As shown in Figure 1, we can align the reads from scratch (de novo assembly); usually by way of overlapping the reads until they form a continuous long read called a contig. The contigs are then overlapped in the same way until they eventually become a full genome.
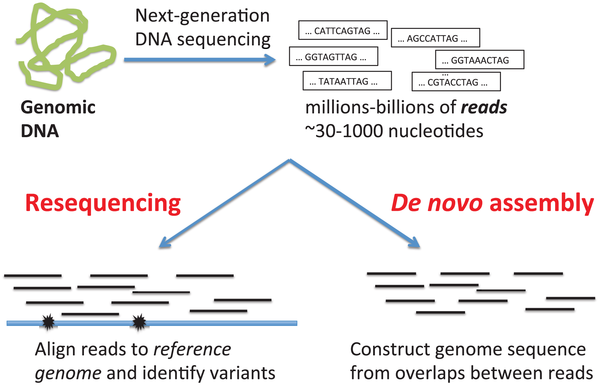
Click here to enlarge the image
Figure 1 – There are two types of reads assembly methods. Either the reads are aligned by overlapping all the reads, until contigs and eventually, a complete reference genome is created. Alternatively, a reference genome may already exist. We would then just align the newly sequenced reads by comparing them to the reference genome. Usually, this method is used to compare the reads, to find variations which exist in our sample compared to the reference genome. Source: PLoS Computational Biology.
A reference genome of a species is therefore a genome that was constructed in this way, by using the sequenced reads of a member of that species. This is normally stored in a FASTA file.
The second method involves using an already available reference genome as a guide, to align the reads of our sample more accurately (resequencing). But the ultimate aim of resequencing is to compare our reads to the reference genome and to find variations.
In Figure 2, the reference genome shows that there is a nucleotide “T” at a particular position. However, the aligned read from your sample shows a “C”. This is a possible mutation. But this is definitely not a process that you would want to complete manually if you have thousands or billions of reads.
 C. Details in the main text”>
C. Details in the main text”>
Click here to enlarge the image
Figure 2 – When there is a different nucleotide at a particular position in your aligned reads, versus the reference genome, then that is considered a mutation. Here the reference genome contains a T at that position, while your read contains a C. Adapted from Your Genome.
Fortunately, the tools required for doing alignments already exist. For short reads (< 300 bp long) produced by a sequencer such as Illumina, we can align the reads to the reference genome using an aligner called BWA-MEM. For longer reads (> 10kb long) produced by a sequencer like Oxford Nanopore, we could align using minimap2.
For resequencing, these tools always require a reference genome (FASTA file) and the FASTQ files containing your reads. You may also need to submit files containing the locations of the primer and adapter sequences that your tool needs to take into consideration when doing the alignment, or which you can use to do prior trimming of your reads before you start the alignment/mapping process. This Difference Between article contains further information on the difference between the two sequences.
Making sense of genomic data: COVID-19 web-based bioinformatics


Making sense of genomic data: COVID-19 web-based bioinformatics
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




