
Variant calling and annotation
Introduction
Identifying genomic variants can play an important role in scientific discovery as exemplified by the ongoing SARS-CoV-2 global surveillance. Variant calling involves identifying single nucleotide polymorphisms (SNPs), and small (< 50 bp) insertions and deletions (indels) from various input data e.g. whole-genome sequence data. Herein, SNPs and indels are collectively referred to as small nucleotide variations (SNVs).
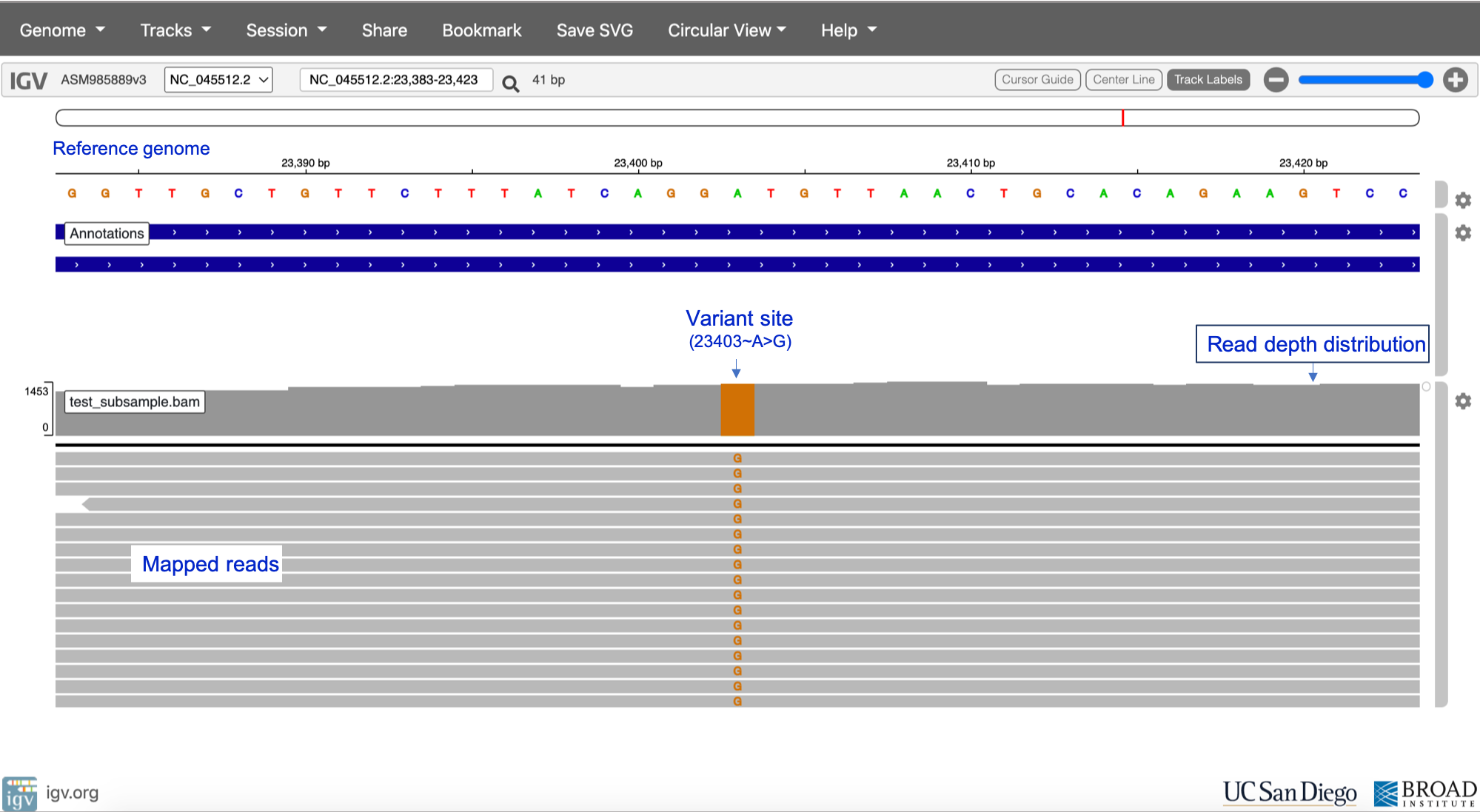
Since NGS-generated reads aligned to the reference genome can be visualised using tools such as the Integrative Genomics Viewer (IGV; https://igv.org/app/), it is possible to visually inspect sites that have potential variants as shown in Figure 1. However, visual inspection of variants would be intractable for the whole genome, hence the need for variant calling tools.
Click here to enlarge the image
Figure 1 – IGV visualisation of a SARS-CoV-2 variant site (with respect to the NC_045512.2 reference) in a simulated sample.
Commonly used variant calling tools include the following;
- BCFtools
- Genomic Analysis Toolkit (GATK) HaplotypeCaller
- DeepVariant
- Strelka2
- SAMtools
- GraphTyper
- Nanopolish
Major computational steps in variant calling are outlined in Figure 2. For variant calling from short-read data (e.g. Illumina), we recommend using GATK HaplotypeCaller, DeepVariant or GraphTyper. DeepVariant has also been validated as highly accurate when using PacBio (long-read) data as input. Nanopolish is the recommended tool for Oxford Nanopore data. For an in-depth benchmark of some of the variant callers above, please see the paper by Barbitoff and colleagues.
 Identifying candidate variant sites > Ascertaining reference and alternate alleles > Assigning sample genotypes i.e. determining whether alleles are homozygous or heterozygous > (For SARS-CoV-2, all variant and reference alleles should be homozygous (i.e. single-stranded genome) > Variant quality control > Output = VCF file”>
Identifying candidate variant sites > Ascertaining reference and alternate alleles > Assigning sample genotypes i.e. determining whether alleles are homozygous or heterozygous > (For SARS-CoV-2, all variant and reference alleles should be homozygous (i.e. single-stranded genome) > Variant quality control > Output = VCF file”>
Click here to enlarge the image
Figure 2 – High-level overview of the variant calling process. BAM – Binary Alignment Map. VCF – Variant Call Format.
It is important to note that different variant callers have different underlying algorithms for detecting and genotyping variants, as well as different approaches for variant quality control and filtering.
Joint discovery vs single sample variant calling
During variant calling, it might be necessary to perform joint discovery rather than single-sample variant calling. In single sample calling, sample BAM files are analysed individually, and individual variant call-sets are combined in downstream steps. However, during joint calling, SNVs are called simultaneously across all sample BAMs, thus generating a merged call-set for the entire cohort. Joint variant calling provides advantages such as facilitating filtering out false positive SNV calls, greater sensitivity for low-frequency variants and enhancing the distinction between homozygous reference sites and sites with missing data. For more insight into the advantages of joint variant calling, please see the GATK article. Although joint variant discovery offers these advantages, there is still a lot of value in performing single sample variant calling – however, the accuracy of the variant calls would depend on multiple factors e.g. the quality of the input data, alignment tool used and the variant caller.
The Variant Call Format
A summary of key components in the Variant Call Format can be found at this resource page. More detailed format descriptions are discussed by Danecek and colleagues and in at SAM tools resources. Figure 3 shows a screenshot of an example VCF for a SARS-CoV-2 sample.

Click here to enlarge the image
Figure 3 – Example VCF output (simplified) after running a variant calling pipeline. The variant NC_045512.2.g.8393G>A is a SNP, while variants NC_045512.2.g.6512AGTT>A and NC_045512.2.g.A>AGCCAGAAGA are deletion and insertion variants, respectively.
Variant annotation
Variant annotation is the process of assigning additional information to mutations called from sequence data – this information could include the effect of a variant on protein structure and function, frequency of the variant (if existing) in well-studied populations, variant identifiers or position of the variant in the context of possible transcripts, proteins, overlapping genes etc. ( Figure 4).

Click here to enlarge the image
Figure 4 – Example of a VCF file output after annotation by snpEff.
The Ensembl Variant Effect Predictor (VEP) and snpEff are the most popular tools used for variant annotation. Both VEP and snpEff are commonly used for annotating variants in the human genome, but they have been adapted to facilitate the annotation of SARS-CoV-2 mutations since the start of the COVID-19 pandemic. There is a dedicated Ensembl VEP for SARS-CoV-2, while the snpEff version(s) for SARS-CoV-2 can be run via the Galaxy web platform.
Have you used any of these tools for variant calling and/or annotation? If you haven’t used the tools before, are you interested in trying them now that you’ve learned how they work? Please use the discussion area to relate your experiences.
Making sense of genomic data: COVID-19 web-based bioinformatics


Making sense of genomic data: COVID-19 web-based bioinformatics
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




