
Interpreting the Sanger sequence data
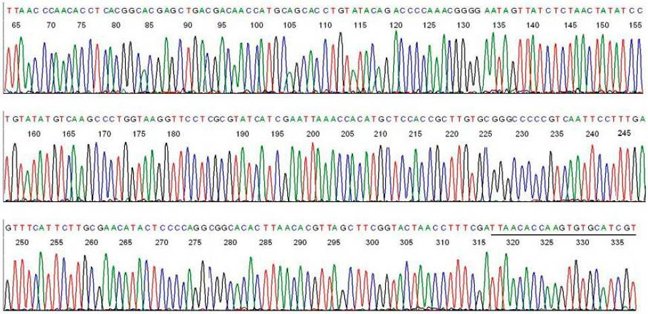
A typical electropherogram from a single PCR product is shown below. Each peak represents a single nucleotide in the DNA sequence, and each nucleotide has a different colour; A is green, T is red, C is blue and G is black.
 From
From The average PCR product contains 200 nucleotides of sequencing, and the maximum length that can be sequenced by the Sanger method is about 600 nucleotides. The amount of data generated by Sanger sequencing for each patient therefore varies depending on the size of the gene being sequenced. Sequencing of a relatively small gene such as HNF1A generates over 2500 nucleotides of sequencing data per patient, and since a mutation can occur at any of the nucleotides within the electropherogram, each nucleotide requires checking in turn for the presence of a mutation.
Sanger sequencing analysis is performed on a comparative basis, where the patient’s electropherogram is compared against an electropherogram from a DNA sample without a mutation. Any observed differences between the two traces are recorded and analysed for their potential pathogenic effect on the protein.
Historically this was performed by a visual comparison of each nucleotide peak in the two traces, but this is time consuming, prone to error and cannot meet the workload and quality demands of today’s modern, high throughput diagnostic laboratory.
Today, sequencing analysis is performed by software that can perform comparative analysis of tens of thousands of nucleotides within seconds. The software automatically detects mutations and provides a description of the mutation at the DNA and protein level with a high degree of accuracy and sensitivity.
Visual inspection: time consuming and error prone 
Software analysis: fast and accurate 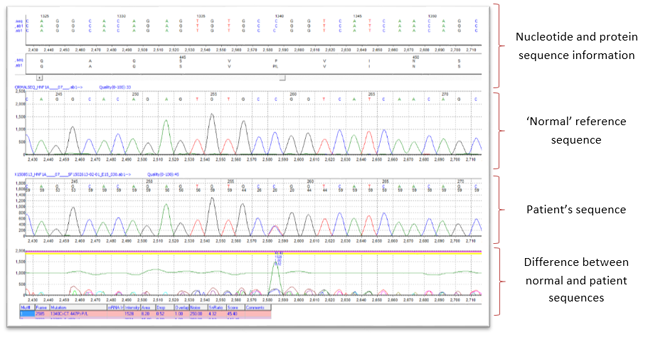 Created by Mutation Surveyor v 4.0.6 SoftGenetics, State College PA USA 16803
Created by Mutation Surveyor v 4.0.6 SoftGenetics, State College PA USA 16803
What was the result of the Sanger sequencing analysis of Dan’s HNF1A gene? The output from the sequencing analysis software can be seen in the picture above. It shows Dan’s DNA sequence with a heterozygous missense mutation in exon 7 of the HNF1A gene. The mutation is the substitution of a C nucleotide with a T nucleotide on one copy of his HNF1A gene (indicated by the presence of a C and T peak at nucleotide position 1340 in Dan’s DNA sequence). This results in a change in the amino acid (from proline to leucine) at amino acid number 447 in the HNF1A protein sequence.
HNF1A is a transcription factor. Its function is to control the expression of other genes in the beta cell of the pancreas that are involved in insulin production and secretion, including the insulin gene itself. Transcription factors work by binding to the promoters of genes and activating gene transcription (the process of making mRNA copies of the gene to be used for protein synthesis). The promoter is the switch that turns the gene on or off, and the transcription factor is the finger that operates that switch.
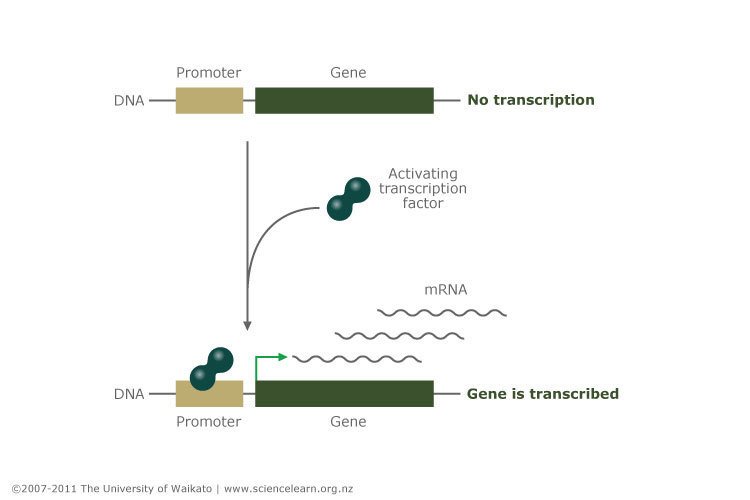 Copyright: University of Waikato. All Rights Reserved. Science Learning Hub
Copyright: University of Waikato. All Rights Reserved. Science Learning Hub
Dan’s mutation occurs within the transactivation domain of the HNF1A protein; the region of the protein responsible for flicking the switch. The mutant HNF1A protein is able to bind to the promoter, but cannot turn on gene expression. This results in defective expression of genes required for insulin production and secretion, which results in insulin deficiency and diabetes. This is a known pathogenic mutation and we can therefore be confident that the result confirms a diagnosis of HNF1A MODY in Dan.
Genomic Medicine: Transforming Patient Care in Diabetes


Genomic Medicine: Transforming Patient Care in Diabetes
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




