
Exome sequencing strategies to identify new disease genes
Using next generation sequencing we can sequence all the ~20,000 genes in the human genome in one single test. This approach is called whole exome sequencing. The coding regions (i.e. the exons) of the genes account for just about 2% of the human genome and it is estimated that approximately 85% of the disease-causing mutations fall within a coding region. Exome sequencing therefore analyses only the portion of our DNA in which the mutation causing a certain disease is most likely to be. Since its introduction exome sequencing has been the most widely used approach to identify new disease-causing genes.
Even though whole exome sequencing only analyses 2% of the genome, ~200 novel/rare variants would be identified in each individual. It would therefore be impossible to look at all of them one-by-one to identify the new disease gene.
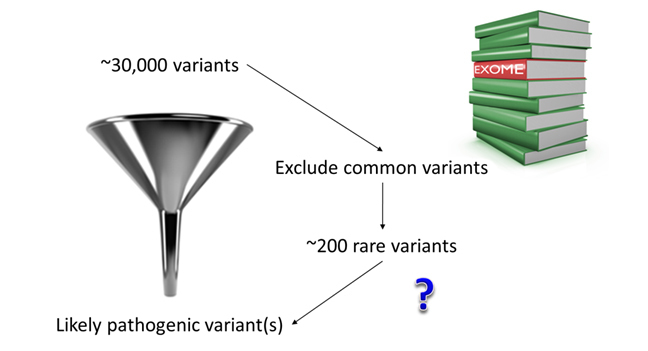
To be sure to recognise the new gene among the hundreds of variants of no clinical significance resulting from whole exome sequencing it is necessary to define a clear strategy. Here are some of the strategies that have been proven most successful so far:
Exome trio strategy
Let’s take the example of Sam. Sam was diagnosed with diabetes at birth. He was born small for gestational age (1385g at 37 weeks). He also had multiple heart defects that needed surgical correction and imaging of his abdomen showed that he had no pancreas at all. Sam’s parents were not known to be related and no one in his family had diabetes or heart defects. This would be Sam’s pedigree:
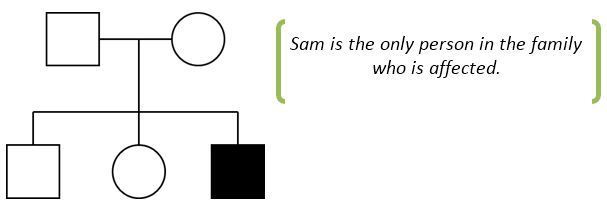
In this example it is likely that the mutation has arisen spontaneously in the patient and has not been inherited by either unaffected parent (geneticists call this a de novo mutation).
We can use exome sequencing to identify a de novo mutation testing Sam (the patient) and his parents. This way we can exclude all the variants that Sam has inherited from either parent and look at the mutations that are found only in Sam. Using this approach, we typically find between 0 and 4 de novo coding mutations in each patient. In Sam’s case we found only one: a mutation in the GATA6 gene which was found to be the cause of the neonatal diabetes, absence of the pancreas and heart malformations in Sam. After finding this mutation in Sam, GATA6 was recognised as being the gene most frequently mutated in patients who are born without a pancreas (a condition called pancreatic agenesis).
Shared phenotype strategy
Sometimes there are multiple patients who clearly have the same very rare syndrome that is likely to have a monogenic basis. In these cases the disease-causing gene can be identified by performing exome sequencing on multiple affected individuals and looking for either the same genetic variant or different variants within the same gene. This strategy led us to identify the genetic cause of Tom’s rare syndrome.
Earlier this week you met Tom who has MDP syndrome (Mandibular hypoplasia, Deafness and Progeroid features). He is the only affected person in his family and it was likely that the disorder was caused by a de novo mutation. You’ll see a striking similarity in both Tom’s facial features and fat distribution with those of a second patient who had also been diagnosed with MDP syndrome. Both consented to exome sequencing so we embarked on a search for variants in a shared gene. In fact we found the same de novo mutation in both patients: a deletion of a single amino acid in the POLD1 gene. The same mutation was identified in two additional patients with MDP syndrome, proving that this particular POLD1 gene mutation is the cause of Tom’s and the other patients’ unique condition.
Finding new genetic causes in consanguineous families
In Week 2 we have introduced you to the different patterns of inheritance of monogenic diseases. According to these models, patients who are born to related parents are at increased risk of inheriting a recessive disease. Therefore in these cases, even if the parents are not affected, it is unlikely that a de novo mutation is the cause of the disease. Sometimes, in families affected with a recessive condition, you could find that there are multiple patients affected in the same generation.
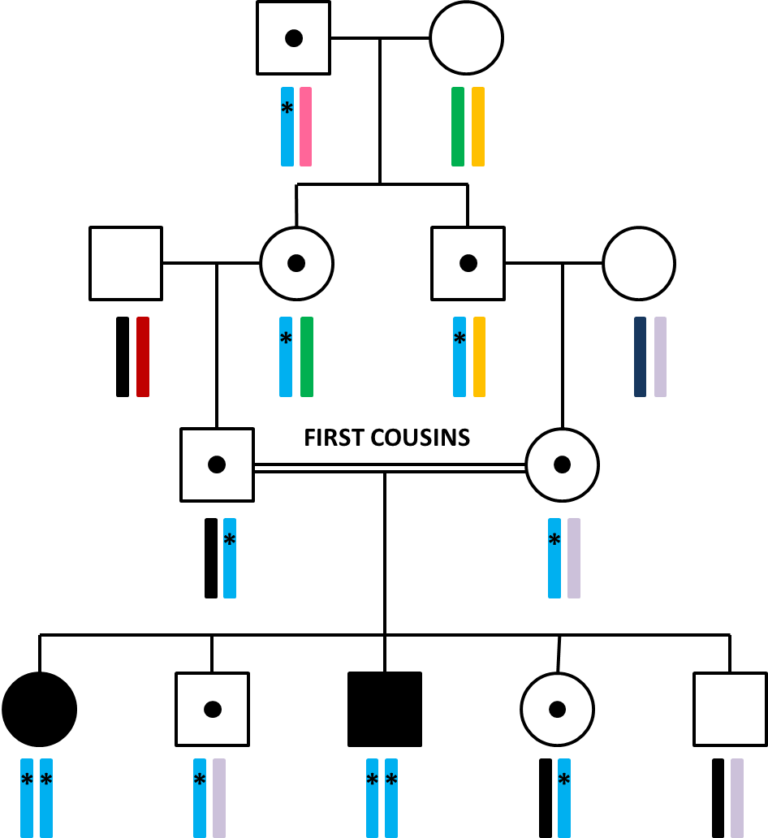
Depending on the relatedness of the parents, the affected patient could have hundreds of potentially pathogenic homozygous variants. How shall we proceed in these cases?
A useful tool to prioritize variants in patients born to consanguineous (related) parents is linkage. In consanguineous families, this would mean analysing both affected and unaffected patients within the same family to identify big regions in which the affected patients are homozygous for all the variants and the unaffected are not. This allows cutting down the potentially disease-causing variants to the ones that fall within the regions of linkage. These regions can then be analysed using exome sequencing. But, even if 85% of the disease-causing variants fall within the exome, sometimes the causal mutation is located outside the coding regions. In these cases, exome sequencing simply isn’t enough… We need genome sequencing!
If you would like to know more about discovering new monogenic subtypes of diabetes, you may find the following articles helpful:
-
GATA6 haploinsufficiency causes pancreatic agenesis in humans, Lango Allen H et al 2011, Nature Genetics
-
An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy, Weedon MN et al 2014, Nature Genetics
Genomic Medicine: Transforming Patient Care in Diabetes


Genomic Medicine: Transforming Patient Care in Diabetes
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




