
Accessing and using sequencing data

Sequencing platforms
There are several different sequencing platforms commercially available. Although we focus here on those developed by Illumina and Oxford Nanopore Technologies (ONT), other available platforms include PacBio by Pacific Biosciences and Ion Torrent by Thermo Fisher Scientific In high-throughput sequencing (HTS), also known as next-generation sequencing (NGS), sequencers create millions of reads. A single read is a computational DNA sequence created from the sequencing of one piece of DNA in the library that is being sequenced. A sequencing library is a pool of DNA fragments with sequencing adapters and barcodes attached. The library pool can be composed of DNA from multiple samples each with their own unique barcode.
It should be noted that whilst Illumina sequencers only sequence DNA molecules, RNA transcriptomic sequence analyses can still be conducted by performing reverse transcription of the sample RNA to DNA before library preparation, whilst Oxford Nanopore sequencers are capable of sequencing either RNA or DNA molecules directly. In the descriptions below, we use DNA as the molecule being sequenced.
Sequence data formats and conversion
Sequencers create millions of reads, which need gigabytes (GB) of computational storage.
The standard format used to computationally store reads is the FASTQ format, which is a text based format for storing nucleotide sequences along with their corresponding quality scores. Quality scores are included for each sequenced base which represent the sequencer’s probability estimate that the base was sequenced correctly.
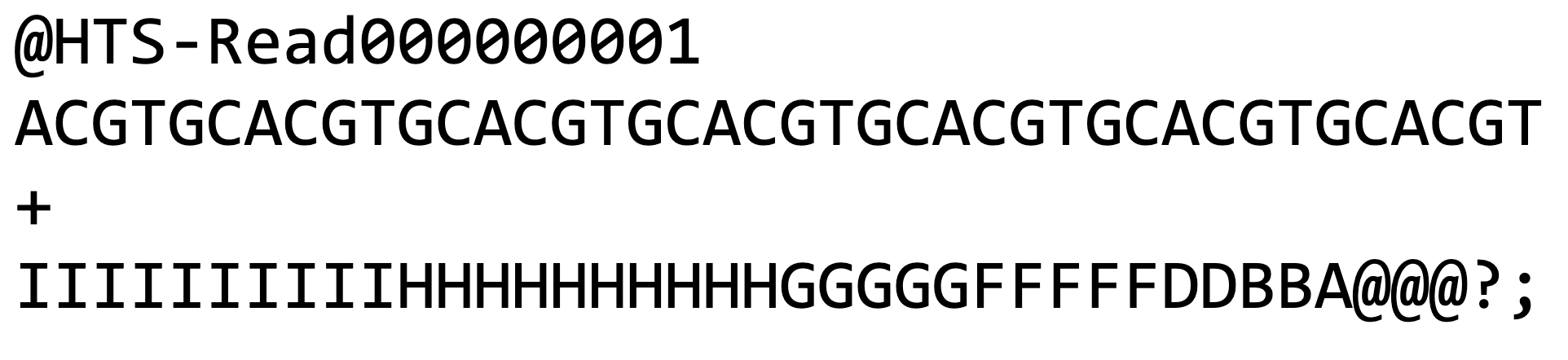
Figure 1 – Example FASTQ read. Four text lines are used to represent each read. The first line is the unique name of the read, the second line is the nucleotide sequence, the third link is the ‘break’ line, and the fourth line is the quality scores (one for each base sequenced). A table of quality score characters and their corresponding error probabilities can be found at Usearch website.
Illumina machines output their raw data in binary base call (BCL) format. Some Illumina sequencers (MiniSeq, MiSeq and NextSeq) provide an option to automatically convert the reads from BCL to the FASTQ format. Whilst other Illumina sequencers (NovaSeq) require separate conversion. This can be accomplished manually by using Illumina’s BCL Convert software (a replacement of the older bcl2fastq software). Alternatively, Illumina has a cloud-based data management and bioinformatics system called BaseSpace (a subscription may be required). Sequencing data is uploaded to BaseSpace which enables real-time monitoring of sequence runs and conversion from BCL to FASTQ formats in the cloud, amongst a host of other functionalities.
All Oxford Nanopore sequencing devices (MinION, GridION, PromethION, etc), use nanopores that correspond to an electrode connected to a channel and sensor chip. The sensor measures the change in the electric current that flows through the nanopore when DNA or RNA molecules pass through. This electric signal data produced from a nanopore read is referred to as a ‘squiggle’ and is stored computationally using the FAST5 format (which is based on the HDF5 format). FAST5 squiggles (electrical reads) can then be converted into the standard FASTQ sequence format by base calling. This can be accomplished using software such as guppy, which can be run manually post-run or run ‘live’ during the run; live base calling works fastest when the computer connected to the nanopore sequencer has a Graphical Processing Unit (GPU). Alternatively, Oxford Nanopore provides a cloud-based data analysis platform called EPI2ME (subscription may be required) where FAST5 data can be uploaded during the sequencing run and base calling can be done live, amongst a host of other functionalities.
A Practical Guide for SARS-CoV-2 Whole Genome Sequencing


A Practical Guide for SARS-CoV-2 Whole Genome Sequencing
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




