
Interpreting Nextclade quality metrics
Nextclade is a free and open-source tool for viral genome alignment, mutation calling, clade assignment, quality checks, and phylogenetic positioning. It can rapidly assess viral genomes by running a series of simple, but useful, calculations that can be used to inspect and classify your newly sequenced genomes or as a pre-submission check prior to depositing them in public databases like NCBI and GISAID. It is developed by the same team behind Nextstrain.
Nextclade is available through a command line or web-based interface available at Nextclade website. You can drag and drop a file containing your SC-2 genomes into your browser window, retrieve sequences from a URL, or paste your FASTA sequences into the text field. Nextclade will detect differences between your sequences and a reference sequence (Wuhan-Hu-1/2019), use these differences to classify your sequences into clades/lineages, and flag probable quality issues in your data. All of the analysis takes place entirely within the browser, with no data ever leaving the computer.
The analytical pipeline includes the following steps:
1) Sequence alignment: Using a custom alignment algorithm, sequences are aligned to the reference genome. 2) Translations: Nucleotide sequences are translated into amino acid sequences. 3) Mutation calling: Changes in nucleotides and amino acids are found. 4) Detection of PCR primer changes in binding sites 5) Phylogenetic placement: Sequences are placed on a reference tree, and private mutations are examined. 6) Assignment of Clades: Clades are derived from the tree’s parent node. 7) Quality Control (QC): Metrics for quality control are computed.
In this example, we examine 61 SC-2 genomes from Peru that were recently sequenced using an Illumina MiSeq instrument. When you press the “run” button, you will immediately see an overview of the results.

Nextclade uses several quality control indicators to quickly identify problems in your sequences. Bad sequences are red, mediocre ones are yellow, and good ones are white. By hovering your cursor over a sequence’s QC entry, you can get the results of the QC metrics.
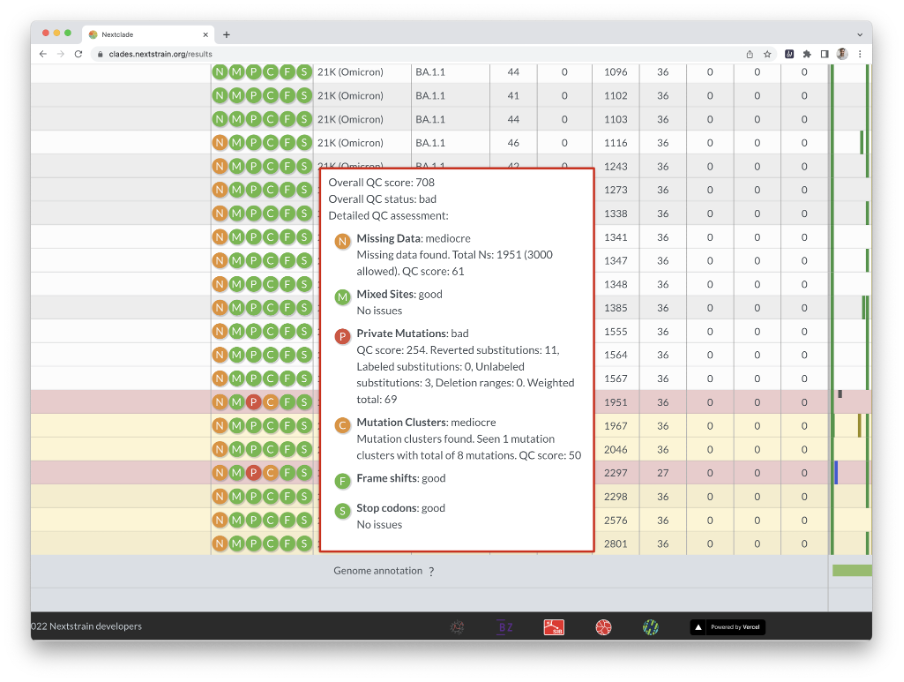
Nextclade also infers the clade/lineage to which a sequence belongs and displays the result in the table. Hovering over the results displays specific information such as the number of mutations, Ns, gaps, insertions, frameshifts, and so on.

The alignment can be seen at the right of the window, with missing data shown in grey. You can readily see how missing data segments are scattered around the genome, whether it’s a few large portions clustered in one location or many small missing parts.
You can zoom in on a gene by clicking on it at the bottom, or by selecting it from the dropdown menu at the top. In sequence view, you can see mutations in a specific gene, which can be shown next to its codon translation.

When Nextclade is done with its analysis, you can download the results in different formats by clicking the download icon in the upper right corner.

References
[Nextclade: clade assignment, mutation calling and quality control for viral genomes[(https://doi.org/10.21105/joss.03773)
Nextstrain FAQs and documentation
A Practical Guide for SARS-CoV-2 Whole Genome Sequencing


A Practical Guide for SARS-CoV-2 Whole Genome Sequencing
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




