
Case study on infection forensics

Phylogenetic trees of bacteria are built using molecular data, often using gene sequences or whole genomes. When investigating a possible outbreak, the phylogeny can be used to explore the evolutionary relationships between isolates from different patients. In this section, we will be studying an outbreak of Mycobacterium abscessus at a cystic fibrosis clinic, which will illustrate how phylogenetics can be useful for understanding transmission routes within a hospital.
M. abscessus causes chronic infections in cystic fibrosis (CF) patients. CF is an inherited, lifelong disorder that causes an excess of sticky mucus secretions in the lungs, making patients highly susceptible to bacterial infections such as M. abscessus. Unlike other CF pathogens such as Pseudomonas aeruginosa, M. abscessus was thought to be non-transmissible, and patients were only able to acquire infections from the environment. In 2010, the genomes of 147 isolates were sequenced in order to investigate an observed increase in cases. These isolates were collected over a period of 4 years, from 31 CF patients attending a single hospital. Before this study, it was unclear whether this increase in cases was a true outbreak, caused by the spread of similar strains, or was more sporadic in nature. Determining the cause of the increase was important for deciding on the most effective infection control measures.
Whole genomes of each isolate were sequenced at the Wellcome Sanger Institute on the Illumina platform and more than 260,000 single nucleotide polymorphisms (SNPs) were identified across the genomes. The SNPs were used to build a maximum likelihood phylogenetic tree.
Using the tree structure
Most isolates from different patients were distantly related with > 100 SNPs. Mycobacteria are thought to have low levels of diversity, so we considered these cases as unrelated and caused by independent acquisition of M. abscessus from the environment. As we had multiple isolates from each patient, we could also see that isolates from the same patient were identical or near-identical, and shared a unique most recent common ancestor (monophyletic).
However, isolates from 11 patients were identical or near identical to other patients’ isolates (see figure), and these isolates were grouped on the phylogenetic tree into two distinct clades. There is no formal definition for a cluster but it typically refers to isolates that belong to the same clade, with relatively small evolutionary distances between them. The two clusters in this dataset indicated to us that there were two concurrent outbreaks.
Within these clusters, it was found that many of the patient’s isolates were paraphyletic, which means that they did not have their own unique common ancestor, and their most recent common ancestor was shared with other patients. This pattern of paraphyly, where genetic diversity of isolates from one patient is nested within another, strongly indicates that there was gradual sequential spread between patients, rather than spread from a single source.
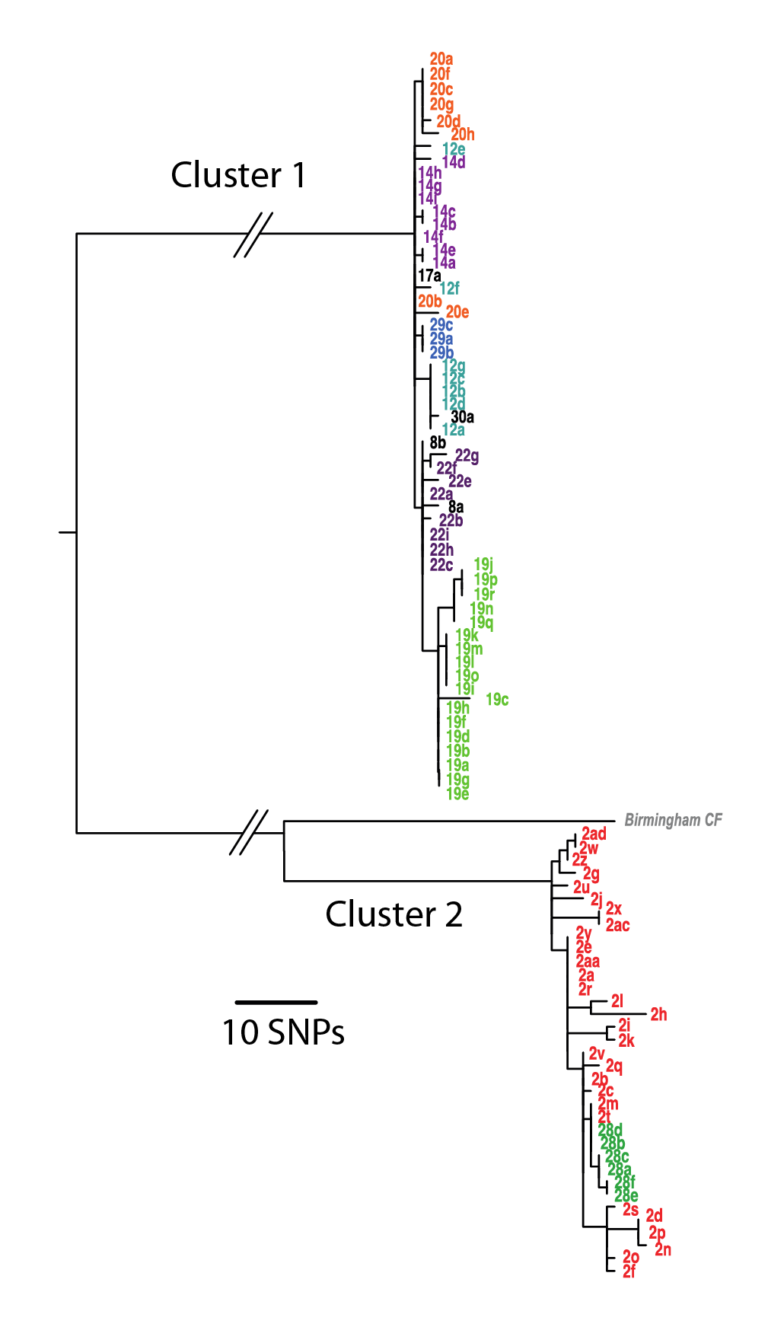
Phylogenetic tree showing relationships between M. abscesses isolates from different patients. (Click image to expand) This figure shows a detailed view of the most closely related bacterial isolates from this study. The numbers indicate different patients, and the letters different isolates from these patients. There is very little genetic distance between the isolates in cluster 1 (look at the horizontal branch lengths). In cluster 2 there is also very little genetic distance between isolates, and all of the isolates from patient 28 cluster within the diversity of isolates from patient 2. This indicates that patient 28 acquired their M. abscessus infection from patient 2. Image © Josie Bryant, 2014
Using genetic distance
If a bacterium has recently been transmitted from one patient to another, the two strains isolated from each patient should be very similar, with the genetic distance between them consistent with the known mutation rate of the bacteria. This is one way of checking that the molecular data is supporting recent transmission and therefore an outbreak.
Different bacterial pathogens can have different mutation rates, some of which have been tested experimentally. However, for many bacteria we do not know the experimental mutation rate, and for the ones where we do have an experimentally derived rate, we don’t know how much this deviates from the rate within patients, where different environmental stressors are likely to be present. To gauge the expected genetic distances within a true outbreak, a common approach is to take multiple isolates per patient over time, and measure the rate of change. For M. abscessus, we found that within patients it evolved at roughly 1 mutation per genome per year, and that the number of SNPs within a patient rarely exceeded 20. In our putative outbreaks, all the genetic distances between patients were lower than 20 SNPs, supporting our conclusion that these were recent transmission events.
Conclusion of the outbreak
Using the whole genome phylogeny, we concluded that there were two distinct transmission chains within the centre, and that these were caused by sequential spread from patient to patient over the past 4 years. The mechanism of transmission was thought to be indirect via respiratory droplets produced by patients, with aerosols left in the air or on objects within a room after treatment. The hospital has now changed its cleaning and air handling mechanisms, and it appears that these transmission chains have halted.
The original research article on this case can be found here Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study in The Lancet
If you are interested in how phylogenetic analysis stands up in court, read about the conviction of a Valencian anaesthetist for infecting his patients with Hepatitis C
http://news.bbc.co.uk/2/hi/europe/6657509.stm © Copyright 2007 BBC
Viral phylogeny in court: the unusual case of the Valencian anesthetist in BMC Biology
Bacterial Genomes: Disease Outbreaks and Antimicrobial Resistance


Bacterial Genomes: Disease Outbreaks and Antimicrobial Resistance
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




