
Introduction to phylogenetic trees
What are phylogenetic trees?
A phylogeny, also known as phylogenetic tree, depicts estimated evolutionary relationships between taxa – these can be species, strains or even genes.
In the context of infectious diseases epidemiology, phylogenetic trees are commonly used to define evolutionary relationships between strains of the same bacterial species. This is possible because bacteria reproduce clonally.
During clonal reproduction, bacterial progenitor cells replicate their DNA at high fidelity. Despite this, random errors in DNA replication may still occur, resulting in a clonal progeny that will inherit these genetic replication ‘errors’ (i.e. mutations) in their DNA and may not be strictly identical to their progenitor cells. Bacterial strains that have recently originated from the same progenitor cell are thus expected to share identical genomes, or have diverged at most by only a few genetic differences (mutations). The number and pattern of shared mutations between bacterial strains can be used to reconstruct their genealogical and evolutionary relationships.
On a phylogenetic tree, isolated bacterial strains are depicted on the tips (or leaves) of the tree (i.e. taxa), whereas the internal nodes of the tree denote their hypothetical ancestors. Nodes and taxa are connected by branches, the length of which represent genetic distances between connected groups. Groups of bacterial strains (taxa) that share the same common ancestor form a monophyletic group (also known as clade). A group of strains that descends from a common ancestor, but does not include all descendants, is called paraphyletic.
How are phylogenetic trees reconstructed?
Today almost all phylogenetic trees are inferred from molecular sequence data, most often from DNA sequences. This is because DNA is an inherited material; it can easily, reliably and inexpensively be extracted and sequenced; and DNA sequences are highly specific to bacterial species and strains.
The application of whole-genome sequencing now makes it possible to ‘read’ the DNA sequence of the whole bacterial chromosome, which provides the ultimate level of resolution possible to discriminate between closely related strains. The figure below illustrates the common workflow to generate multiple DNA sequence alignments from a collection of bacterial strains. Generally, bacterial DNA is extracted from a single colony picked from culture plates (referred as to ‘isolate’), followed by library preparation and whole-genome sequencing using rapid benchtop sequencers. Raw sequence data generated by sequencers are processed using bioinformatic and genomic pipelines, which generally involve mapping (aligning) the generated short reads to a reference genome to reconstruct the isolate’s DNA sequence along the whole bacterial chromosome. Mapping the short reads of multiple sequenced isolates to the same reference genome allows the creation of multiple sequence alignments. Multiple sequence alignments are the first and critical point from which phylogenetic trees can be re-constructed.

Polymorphic sites (that is, nucleotide positions that are variable across multiple strains) in multiple alignments are used to infer evolutionary relationships, whereas monomorphic sites (nucleotide positions with the same DNA base) are generally ignored. The figure below shows an example of a simple multiple alignment of eight sites from four strains, which include monomorphic (squared) and polymorphic sites. Genetic changes in the phylogenetic tree are showed as coloured vertical rectangles on the branch where they originated. The identification of genetic changes (alleles) that are unique and common to multiple taxa (strains) are used to group them into monophyletic groups (clades) in a hierarchical manner (see example below) with the goal of constructing the most plausible genealogical relationships between strains and clades.
How are phylogenetic trees interpreted?
The preferred interpretation of a phylogenetic tree is as a depiction of lines of descent. That is, trees communicate the evolutionary relationships among strains and clades. Under this interpretation, internal nodes on a tree are taken to correspond to bacterial strains that existed in the past (ancestral) but could not be sampled.
Phylogenies are commonly mis-interpreted when read along the tips. Instead, the correct way to read a tree is as a set of hierarchically nested groups (clades).
In the tree above, strain C is more closely related to strain B than it is to strain A. This is inferred by tracing the ancestor of strains (depicted as internal nodes) using the branch structure (i.e. topology) of the tree. Relatedness should be understood in terms of common ancestry— the more recently strains share a common ancestor, the more closely related they are.
How are phylogenetic trees used in infectious diseases epidemiology?
Phylogenetic trees are commonly used to identify where person-to-person transmission occurs; to identify the sources and study the transmission routes of outbreak and epidemic clones; and to determine whether bacterial clones are restricted to specific hosts and settings or, on the contrary, able to circulate among multiple ones.
A common phylogenetic method used to study how bacterial characteristics (traits) evolved is ancestral state reconstruction. In the example shown below, strains on the same tree are labelled based on the presence of different traits. Arrows indicate what internal node (ancestor) in the tree most likely changed (lost or gained) such a trait. Bacterial traits we may be interested in reconstructing include: geographical location – to then identify movement between regions (transmission events; colonising or infecting host – to enable us to identify host jumps; and antibiotic susceptibility – to enable us to identify evolution of AMR. The emergence and spread of individual mutations, genes and mobile genetic elements can also be reconstructed in a bacterial phylogeny using this method (see figure below).
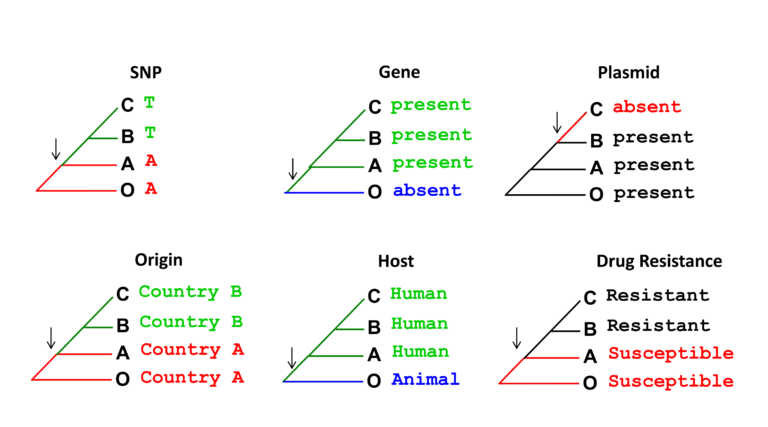
In later steps this week, we will learn how phylogenetic trees were used in real epidemiological investigations to determine the evolution and source of multi-drug resistance bacterial clones.
Bacterial Genomes: Antimicrobial Resistance in Bacterial Pathogens


Bacterial Genomes: Antimicrobial Resistance in Bacterial Pathogens
Reach your personal and professional goals
Unlock access to hundreds of expert online courses and degrees from top universities and educators to gain accredited qualifications and professional CV-building certificates.
Join over 18 million learners to launch, switch or build upon your career, all at your own pace, across a wide range of topic areas.




